
INFO MEDICHE
LE ARITMIE GENETICHE
Gli sviluppi degli ultimi anni nel campo della genetica umana, hanno avuto, in ambito cardiologico, un impatto di fondamentale importanza. Dopo l’identificazione delle basi delle malattie monogeniche del cuore, l’interesse dei cardiologi si è rivolto alla biologia molecolare.
Le nuove conoscenze acquisite hanno permesso di chiarire alcune cause delle morti improvvise per arresto cardiaco in età giovanile in individui senza evidenza di malattia cardiaca strutturale e di porre le indicazione ad interventi terapeutici o protettivi differenti (terapie farmacologiche, impianto di ICD).
- La Sindrome del QT Lungo
- La Sindrome di Brugada,
- LaTachicardia Ventricolare Polimorfa Catecolaminergica
- La sindrome del QT corto
- La cardiomiopatia aritmogena del ventricolo destro
Sono patologie aritmogene ereditarie, la cui eziopatogenesi è individuabile in un’alterazione strutturale, su base genetica, dei canali ionici a livello cardiaco.
Il cuore dei soggetti geneticamente affetti presenta una predisposizione allo sviluppo di aritmie ventricolari. Lo spettro delle manifestazioni di tali sindromi, è estremamente variabile: dalla completa assenza di segni clinici ed ECG grafici normali, a manifestazioni cliniche di particolare gravità.
Pertanto , l’identificazione del difetto genetico contribuisce ad un migliore inquadramento della patologia con la possibilità di integrare la terapia tradizionale con l’introduzione accorgimenti terapeutici gene-specifici, ma anche e soprattutto alla diagnosi nei soggetti apparentemente sani , ma con un rischio aritmico superiore alla popolazione generale.
LA SINDROME DEL QT LUNGO
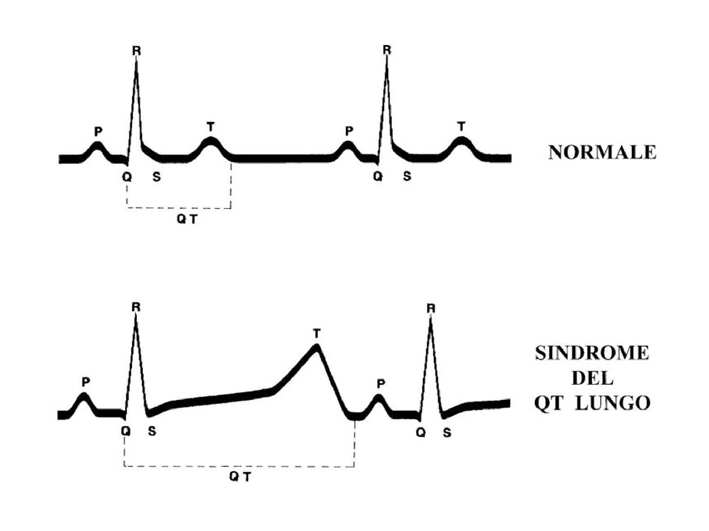
La Sindrome del QT Lungo (LQTS) è una malattia che colpisce la trasmissione elettrica all’interno del cuore. Per ogni battito cardiaco infatti è necessario che vi sia un preciso passaggio di cariche elettriche veicolate da sodio e potassio. L’ingresso di SODIO nelle cellule miocardiche avvia il battito cardiaco mentre l’uscita di POTASSIO riporta il cuore in condizioni di riposo e lo rende pronto per l’attivazione del battito successivo. La LQTS si verifica quando vi sono alterazioni in queste correnti di ioni e, di conseguenza, il tempo richiesto alla componente elettrica del cuore per ritornare nelle condizione di riposo, è maggiore del normale. L’abnorme durata di quest’ultima fase si presenta all’elettrocardiogramma (ECG) con un prolungamento dell’intervallo QT. Nella figura si può vedere che il QT è l’intervallo di tempo compreso tra l’onda Q e l’onda T della registrazione elettrocardiografica. Risulta pertanto chiaro che il nome della malattia “Sindrome del QT Lungo” (abbreviato in LQTS) deriva dall’intervallo QT dell’elettrocardiogramma che è più lungo del normale nei soggetti affetti dalla malattia.
La LQTS può essere di origine genetica (cioè causata da un gene anormale) o acquisita (secondaria a perdite di sali o all’assunzione di farmaci): in entrambi i casi i pazienti affetti sono predisposti ad improvvisa insorgenza di aritmie ventricolari pericolose (tipica è la torsione di punta) che si possono manifestare come episodi sincopali o, nei casi più gravi possono causare morte improvvisa conseguente ad un arresto cardiaco. Esistono anche casi di LQTS in cui la causa non è ne’ genetica ne’ acquisita.
La prevalenza della LQTS non è conosciuta con esattezza, ma si stima attualmente che sia presente in circa 1 persona su 2000: secondo questo dato potremmo concludere che in Italia vi sono circa 30.000 persone affette. La malattia colpisce indifferentemente tutte le razze ed i gruppi etnici; non è noto, attualmente, se vi sia correlazione tra la differenza di razza e la presenza di mutazioni diverse.
(qui vorrei approfondire parlando anche di: tipi di LQTS genetici – valori di riferimento – terapie)
LA SINDROME DI BRUGADA
La Sindrome di Brugada, identificata come entità clinica nel 1992, è una patologia caratterizzata da specifiche alterazioni dell’elettrocardiogramma ed un rischio di insorgenza di aritmie che possono condurre a morte improvvisa. E’ una malattia aritmogena su base genetica che provoca una instabilità elettrica del cuore che lo rende suscettibile all’insorgenza di aritmie in alcune circostanze.
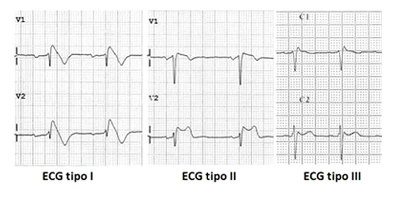
Figura: tre possibili ECG di uno stesso paziente con sindrome di Brugada. Solo il tipo I, ottenuto tramite l’infusione di ajmalina o flecainide, è considerato diagnostico della malattia.
La mancanza di dati completi sulle basi genetiche della malattia rende difficile una stima precisa della prevalenza della sindrome di Brugada. Tuttavia i dati ottenuti considerano la frequenza delle alterazioni elettrocardiografiche della malattia su ampi campioni della popolazione.
Se si considerano complessivamente tutti i lavori scientifici pubblicati al riguardo si osserva una prevalenza media dello 0.10%, ovvero di una persona ogni 1000 abitanti. Se questa figura dovesse essere confermata anche negli studi futuri, la sindrome di Brugada si collocherebbe tra le patologie genetiche a più alta prevalenza nella popolazione.
(qui vorrei inserire un collegamento che parla degli ultimi sviluppi della ricerca su Brugada)
LA TACHICARDIA VENTRICOLARE POLIMORFA CATECOLAMINERGICA
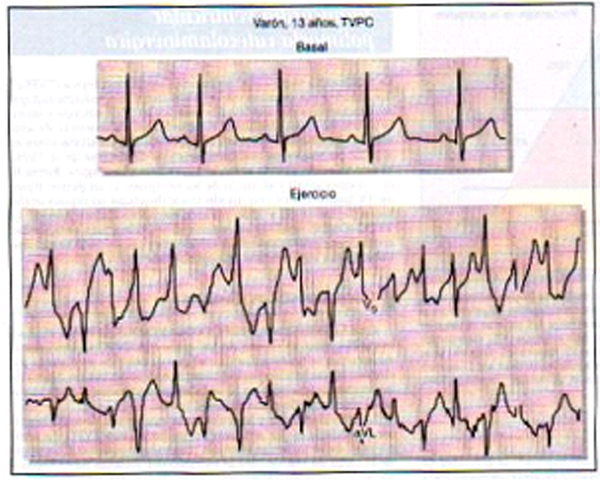
La Tachicardia Ventricolare Polimorfa Catecolaminergica (CPVT) è un’aritmia cardiaca su base genetica che in condizioni di stress fisico o emotivo può provocare perdita di conoscenza o morte improvvisa a causa di una forma di aritmia chiamata “tachicardia ventricolare”. La CPVT è stata identificata come entità clinica nel 1978, ma pochissimi casi al mondo sono stati descritti. Negli anni successivi sono state riportate ulteriori casistiche, ma con un numero relativamente limitato di pazienti. Solo a partire dal 2000, con l’identificazione dei geni di questa malattia si è imparato a conoscerne nel dettaglio le manifestazioni cliniche ed i sottostanti meccanismi.
LA SINDROME DEL QT CORTO
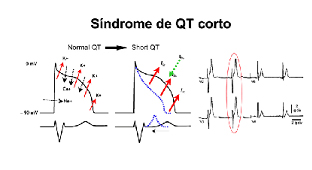
La Sindrome del QT Corto (SQTS) è una malattia che colpisce la trasmissione elettrica all’interno del cuore. Per ogni battito cardiaco infatti è necessario che vi sia un preciso passaggio di cariche elettriche veicolate da sodio e potassio. L’ingresso di SODIO nelle cellule miocardiche avvia il battito cardiaco mentre l’uscita di POTASSIO riporta il cuore in condizioni di riposo e lo rende pronto per l’attivazione del battito successivo. La SQTS si verifica quando vi sono alterazioni in queste correnti di ioni e, di conseguenza, il tempo richiesto alla componente elettrica del cuore per ritornare nelle condizione di riposo, è minore del normale. La minore durata di quest’ultima fase si presenta all’elettrocardiogramma (ECG) con un accorciamento dell’intervallo QT, che sarà inferiore a 340 msec (l’intervallo QT è il tempo compreso tra l’onda Q e l’onda T, che corrisponde al tempo di depolarizzazione e ripolarizzazione: da questo nasce il nome di Sindrome del QT Corto. La SQTS può avere una base genetica e i pazienti affetti sono predisposti ad improvvisa insorgenza di aritmie ventricolari pericolose che si possono manifestare come episodi sincopali o, nei casi più gravi possono causare morte improvvisa da arresto cardiaco. La prevalenza della SQTS non è conosciuta con esattezza, ma si stima attualmente che la malattia sia molto rara.
LA CARDIOMIOPATIA ARITMOGENA DEL VENTRICOLO DESTRO (ARVC)
La Cardiomiopatia Aritmogena del Ventricolo Destro (ARVC) è una patologia aritmogena su base genetica in cui le cellule muscolari cardiache vengono sostituite da tessuto fibroadiposo. Il fenomeno di sostituzione avviene principalmente a carico del ventricolo destro, ma è possibile anche un coinvolgimento del ventricolo sinistro. Il processo di sostituzione fibroadiposa può avere due effetti: causare una elevata predisposizione allo sviluppo di aritmie ventricolari maligne e, più raramente, causare una dilatazione del ventricolo destro.
Nel caso in cui la dilatazione comprometta anche il ventricolo sinistro la malattia può evolvere verso uno scompenso cardiaco. La patologia ha un andamento evolutivo e, perciò, nel tempo, la maggioranza dei pazienti mostra alterazioni dell’ECG ed anomalie strutturali all’ECOcardiogramma ed alla RMN cardiaca. E’ difficile una stima precisa della prevalenza della ARVC, ma è stimato che essa sia di circa 1/5000. La ARVC è considerata una delle principali cause di morte improvvisa giovanile, in particolare negli atleti.
Come fare: